



科学突破:钠离子竟是细胞死亡的主动调控者,而非被动结果
发表时间:2026-04-09
这篇发表于Nature Communications的研究,由上海交通大学医学院钟清团队完成,首次完整阐明钠过载坏死(NECSO)的分子机制:Na?不是细胞死亡的被动结果,而是破坏线粒体能量代谢来引发细胞死亡,Na?作为坏死的主动启动者,而不仅仅是渗透压破坏的晚期参。
一.核心信息速览
1. 研究对象:钠过载坏死(NECSO,Necrosis by Sodium Overload)。
2. 核心启动:小分子NC1持续激活TRPM4通道 → 大量 Na?内流。
3. 核心枢纽:线粒体NCLX(Na?/Ca2?交换体)。
4. 核心结局:线粒体能量崩溃 → Na/K ATPase 失活 → 离子梯度崩塌 → 细胞渗透性肿胀裂解。
5. 科学突破:Na?是主动致死信号,而非仅渗透压效应;明确TRPM4 NCLX 线粒体能量轴是 NECSO 的完整通路。
二.研究背景与核心问题
1. 传统认知 vs 新发现
|
传统观点 |
本研究新观点 |
|
Na?内流是细胞死亡的晚期结果(细胞膜破裂后) |
Na?内流是细胞死亡的主动启动者 |
|
Na?主要通过渗透压失衡导致细胞死亡 |
Na?通过抑制线粒体能量产生主动诱导坏死 |
|
Na?是"被动执行者" |
Na?是"主动调控者" |
2. 团队前期已发现(详细文献解析链接:https://www.enkilife.cn/article/view/0FEA0FA017C3D8763927BA3262E4FFC9):
(1)NC1 激活TRPM4(一价阳离子通道),引发 Na?过载坏死,命名NECSO。
(2)NECSO独立于凋亡、坏死性凋亡、铁死亡、焦亡,不受经典通路抑制剂 / 敲除影响,TRPM4 敲除则完全阻断。
3. 本文待解决关键问题
(1)Na?内流如何导致细胞死亡?
(2)线粒体在 NECSO 中扮演什么角色?
(3)离子稳态与能量代谢如何耦联?
(4)最终膜破裂的执行机制是什么?
三、研究结果
1. 在NECSO中,钠超载在细胞破裂前引发能量耗竭和线粒体离子失衡
(1) 时序证据:Na?内流→早期能量耗竭→后期膜破裂(因果确立)
- NC1 处理 30 min:胞内 Na?显著升高;
- 2–4 h:ATP 急剧下降,PI 阳性 < 10%;
- 18 h:PI 近 100%,LDH 释放更晚→ 能量耗竭早于膜破损,是致死因而非果。
(2) Na?依赖性验证
- 无 Na?培养基(NMDG?替代):NC1 不升高胞内 Na?、ATP 不下降、无细胞死亡;
- 回补 Na?:死亡恢复→ Na?内流是能量崩溃与 NECSO 的必要条件;
- 拯救实验:肌酸、α 硫辛酸(促进 ATP 生成)→ 抑制 NC1 诱导的 LDH 释放→ 能量补充可阻断 NECSO。
-
证明:能量耗竭是 NECSO 的早期核心事件,而非膜破裂后的继发结果。
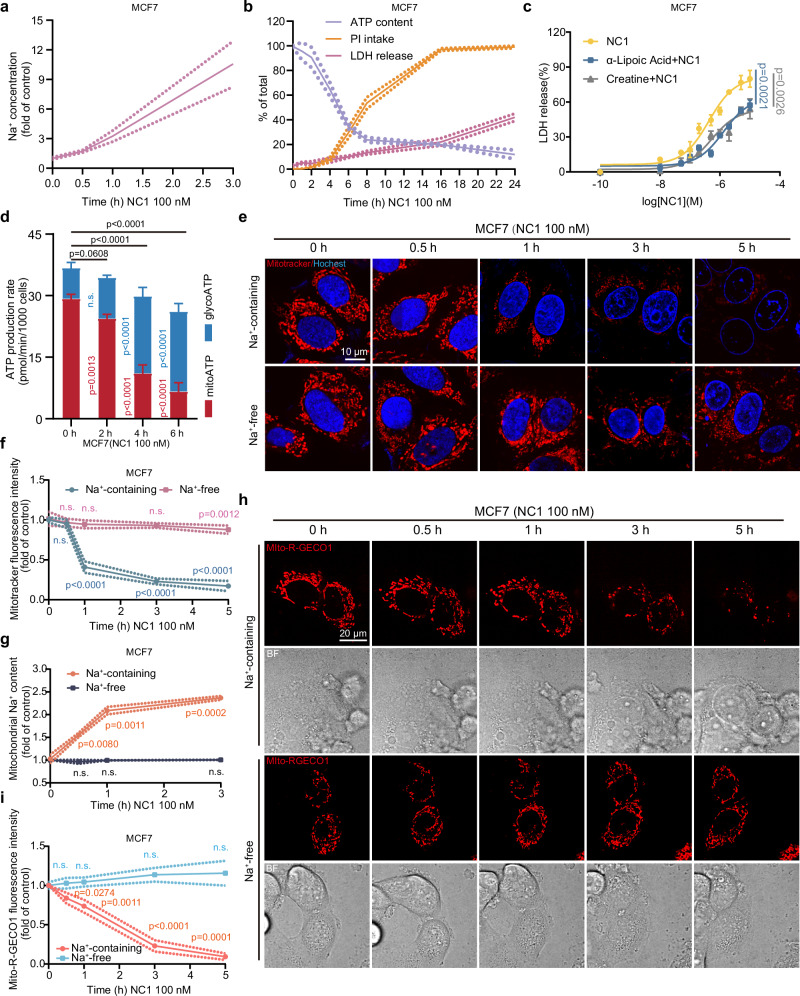
图1. NC1诱导的能量耗竭和线粒体离子失衡依赖于钠离子内流
2. Na?内流直接抑制线粒体能量产生
(1)线粒体结构:NC1 处理后快速肿胀、嵴断裂,无 Na?则不出现;
(2)线粒体离子失衡:
- 胞质 Na?↑→通过NCLX进入线粒体→线粒体 Na?↑;
- 同时 NCLX 交换导致线粒体 Ca2?↓;
(3)代谢与呼吸:
- 氧化磷酸化(OXPHOS)受到抑制,线粒体 ATP(mitoATP)锐减;
- 糖酵解代偿性上升,但总 ATP 仍下降;
- 呼吸链复合体 II+III 活性被高 Na?直接抑制。
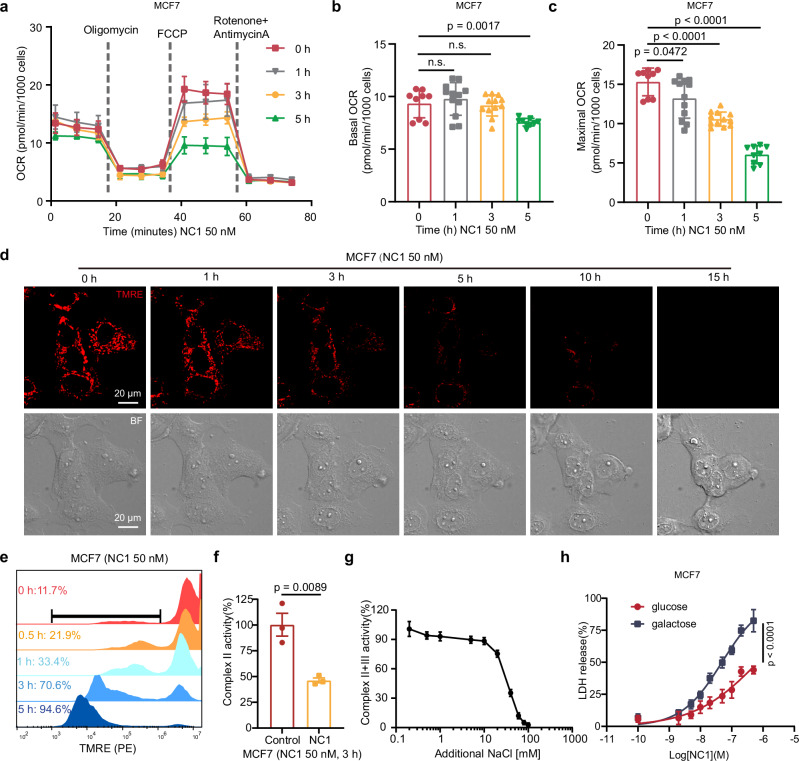
图2. NECSO中线粒体呼吸受到抑制
3. 三羧酸循环在NECSO中被抑制
(1)关键发现:
- TCA循环中间产物积累: 异柠檬酸、α-酮戊二酸、琥珀酸、延胡索酸、苹果酸;
- 时间依赖性: 随NC1处理时间延长而累积;
(2)琥珀酸/延胡索酸比值→进行性升高(0→3小时: ~3倍→~12倍)→证实琥珀酸脱氢酶(SDH/复合体II)功能受抑。
(3)丙酮酸代谢障碍:
- 丙酮酸积累;
- 硫胺素焦磷酸(TPP,PDHc辅因子)积累;
- PDHA1(E1α亚基)Ser293磷酸化增加 → PDHc失活。
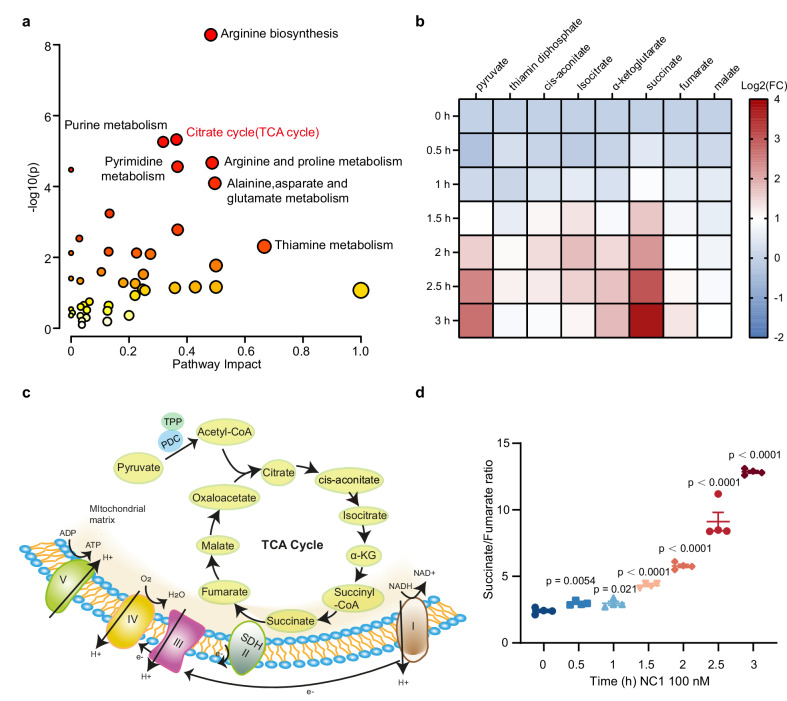
图3. NECSO中TCA循环被抑制
4. 钠超载诱导的线粒体功能障碍依赖于线粒体NCLX
| CGP37157(NCLX抑制剂)实验结果 | |||
| 检测指标 | NC1单独 | NC1+CGP | 效果 |
| 线粒体肿胀(TEM) | 严重 | 明显减轻 | ?保护 |
| 线粒体Na? | ↑↑ | 逆转 | ?阻断 |
| 线粒体Ca2? | ↓↓ | 恢复 | ?逆转 |
| 膜电位(TMRE) | 崩溃 | 维持 | ?保护 |
| 最大呼吸 | ↓↓ | 恢复 | ?逆转 |
| 复合体II+III活性 | ↓↓ | 部分恢复 | ?改善 |
| ATP产生率 | ↓↓ | 恢复 | ?逆转 |
| 细胞死亡(LDH) | ++++ | + | ?显著抑制 |
| 证明: NCLX介导的Na?内流/Ca2?外流是线粒体功能障碍的核心环节。 | |||
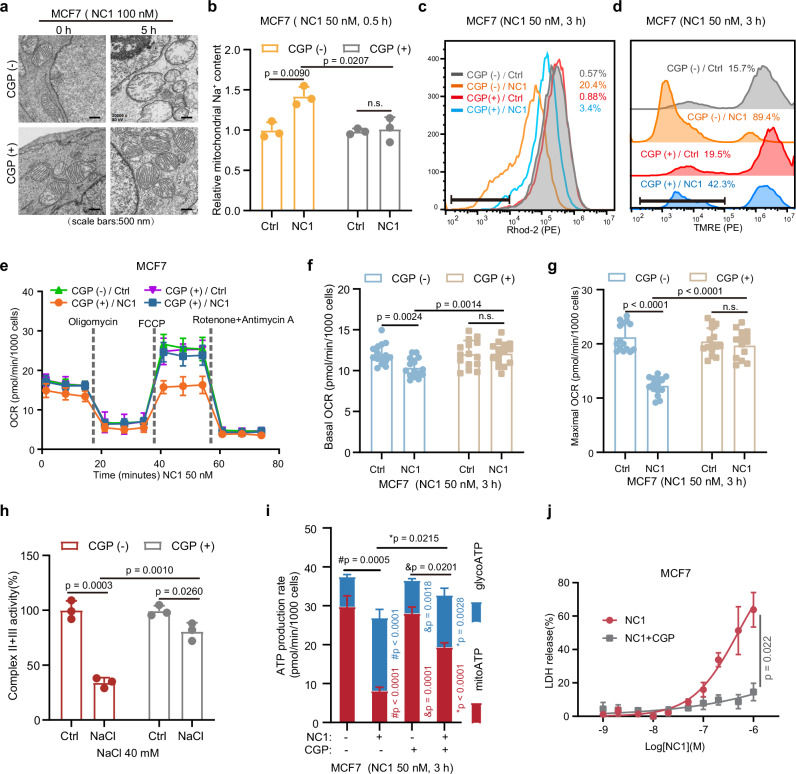
图4. 抑制NCLX可逆转NC1诱导的线粒体功能障碍
5. 能量衰竭→Na/K ATPase 失活→离子梯度崩溃
(1)Na/K-ATPase活性变化:NC1处理1小时后活性开始下降,随时间延长进一步抑制。
(2)Ouabain(Na/K-ATPase抑制剂)效应:
- 加剧NC1诱导的Na?积累、K?流失;
- 极低浓度(10 nM)即可显著促进NC1诱导的细胞死亡;
- 证明Na/K-ATPase抑制是NECSO的关键执行步骤。
(3)Na?-free条件验证:
- 无胞外Na?时,NC1不抑制Na/K-ATPase活性;
- 证实Na?内流是Na/K-ATPase抑制的前提。
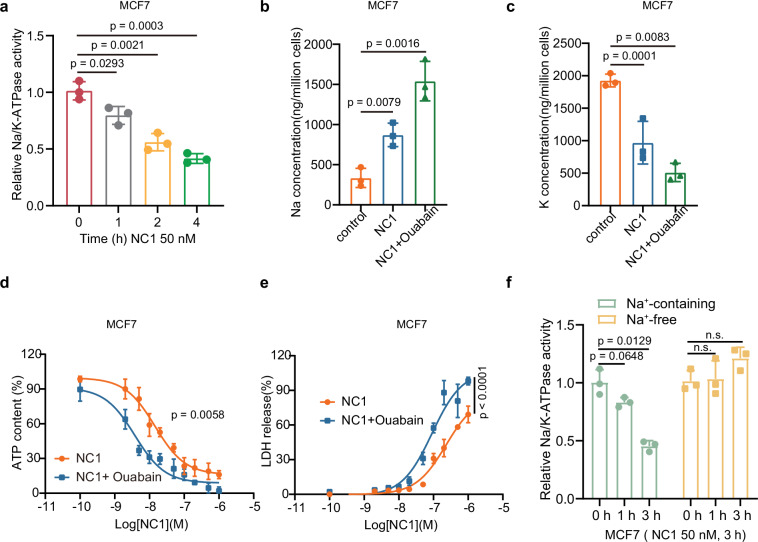
图5. 能量耗竭通过抑制Na/K-ATP酶导致单价阳离子梯度崩溃
6. TRPM4 是 NECSO 中线粒体功能障碍和能量耗竭所必需的![]()
|
TRPM4敲除(TKO)实验结果 |
||
|
检测指标 |
野生型(WT) |
TRPM4-KO |
|
CoroNa Green荧光(胞质Na?) |
NC1后显著↑ |
无变化 |
|
线粒体Na? |
NC1后↓ |
无变化 |
|
线粒体Ca2? |
NC1后↓ |
无变化 |
|
线粒体形态(TEM) |
肿胀 |
正常 |
|
膜电位 |
崩溃 |
正常 |
|
OCR(基础/最大) |
↓↓ |
正常 |
|
复合体II活性 |
↓ |
正常 |
|
ATP产生率 |
↓↓ |
正常 |
|
Na/K-ATPase活性 |
↓ |
正常 |
|
细胞死亡(LDH) |
++++ |
几乎无 |
关键结论:
- NC1不直接作用于线粒体;
- 所有效应依赖于TRPM4介导的Na?内流;
- 建立完整信号链: NC1 → TRPM4 → Na?内流 → NCLX → 线粒体功能障碍。
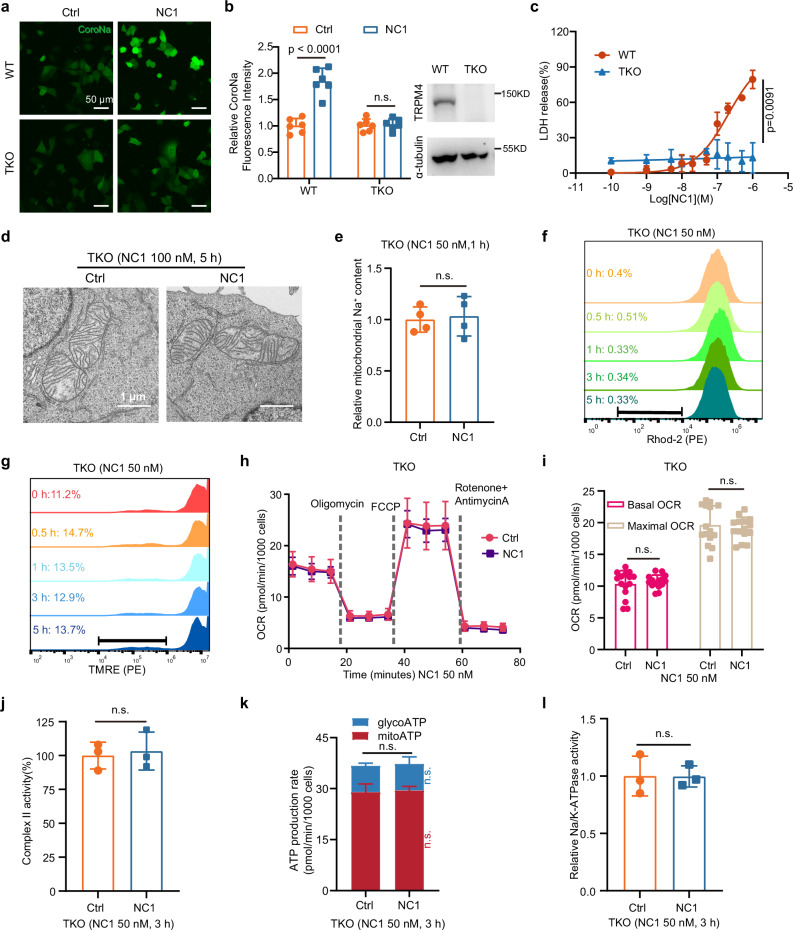
图6. TRPM4是NC1诱导的线粒体功能障碍所必需的
7. 渗透保护剂和AQP4抑制可抑制NECSO
(1)渗透保护剂实验揭示膜孔形成
原理:
- 离子梯度崩溃 → 渗透压升高 → 水内流 → 细胞肿胀;
- 膜形成纳米级缺损 → 大分子可渗漏。
实验结果:
PEG 8K几乎完全阻断NC1诱导的LDH释放、细胞肿胀和PI摄取:证明NECSO中存在大小可变的膜缺损。
渗透保护剂
水合直径
LDH释放抑制
蔗糖
~0.9 nm
部分
PEG 2K
~2.6 nm
较好
PEG 4K
~3.2 nm
更好
PEG 8K
~4 nm
几乎完全抑制
(2)AQP4参与水内流
AQP4抑制实验:
- AER-271(AQP4小分子抑制剂): 显著降低LDH释放;
- AQP4 siRNA: 两种独立序列均显著减少细胞死亡;
- 提示AQP4介导的水内流参与NECSO执行。
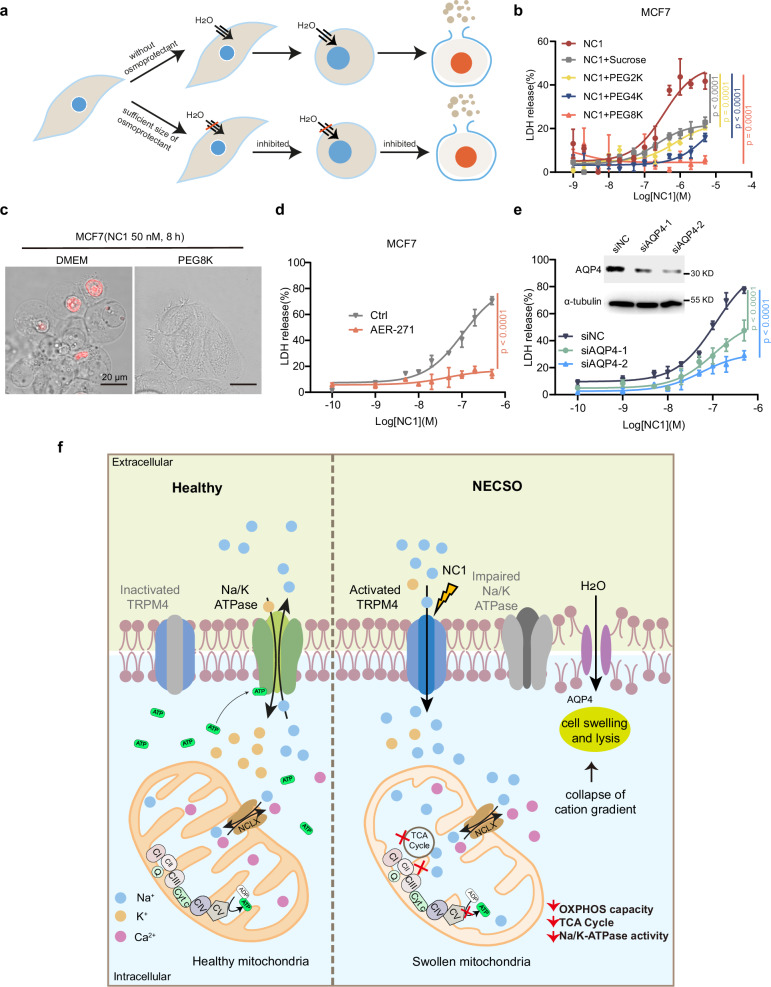
图7. 渗透保护剂和AQP4抑制剂可阻止NECSO中的膜破裂
三、机制总结:NECSO的完整信号通路
该研究表明,钠内流可通过抑制线粒体能量产生来调控细胞死亡。在机制上,该研究揭示了依赖于TRPM4的NC1诱导的钠内流可激活NCLX介导的转运,导致[Na+]升高和[Ca2+]降低。这种扰动会阻碍三羧酸循环、线粒体氧化磷酸化及整体能量生成。因此,钠钾泵的活性受损,最终导致细胞内阳离子梯度崩溃,促进渗透性细胞肿胀和膜破裂。
文献来源:
Sodium disrupts mitochondrial energy metabolism to execute NECSO. Qiao Y, et al. Nat Commun. 2025. [PMID: 41390760]
EnkiLife恩玑生命相关产品:

抗体标记试剂盒
Western Blot全流程实验方案
TSA多重荧光试剂盒
细胞荧光染料
稳转细胞系构建服务